Sunsetting Enforcement Discretion for CLIA-Certified High-Complexity Tests – FDA Presses Ahead With LDT Final Rule
On May 6, 2024, the US Food and Drug Administration (FDA) published its final rule to overhaul FDA’s approach for laboratory-developed tests (LDTs). The final rule – issued seven months after the proposed rule was published1 – amends FDA’s regulations to make clear that in vitro diagnostics (IVDs) include LDTs under the Federal Food, Drug, and Cosmetic Act (FDCA). The final rule also sets forth FDA’s policy to phase in enforcement of the FDCA requirements for most LDTs. FDA will host a webinar on the final rule on May 14, 2024.
Main elements of the final rule
The final rule amends the definition of “in vitro diagnostic products,”2 which states that “[t]hese products are devices as defined under section 201(h) of the [FDCA].” To emphasize that the IVD definition includes LDTs, FDA adds to the end of this definition the phrase “including when the manufacturer of these products is a laboratory.”3
A key component to implementing the final rule is sunsetting FDA’s current LDT enforcement discretion policy4 over the next four years as follows:
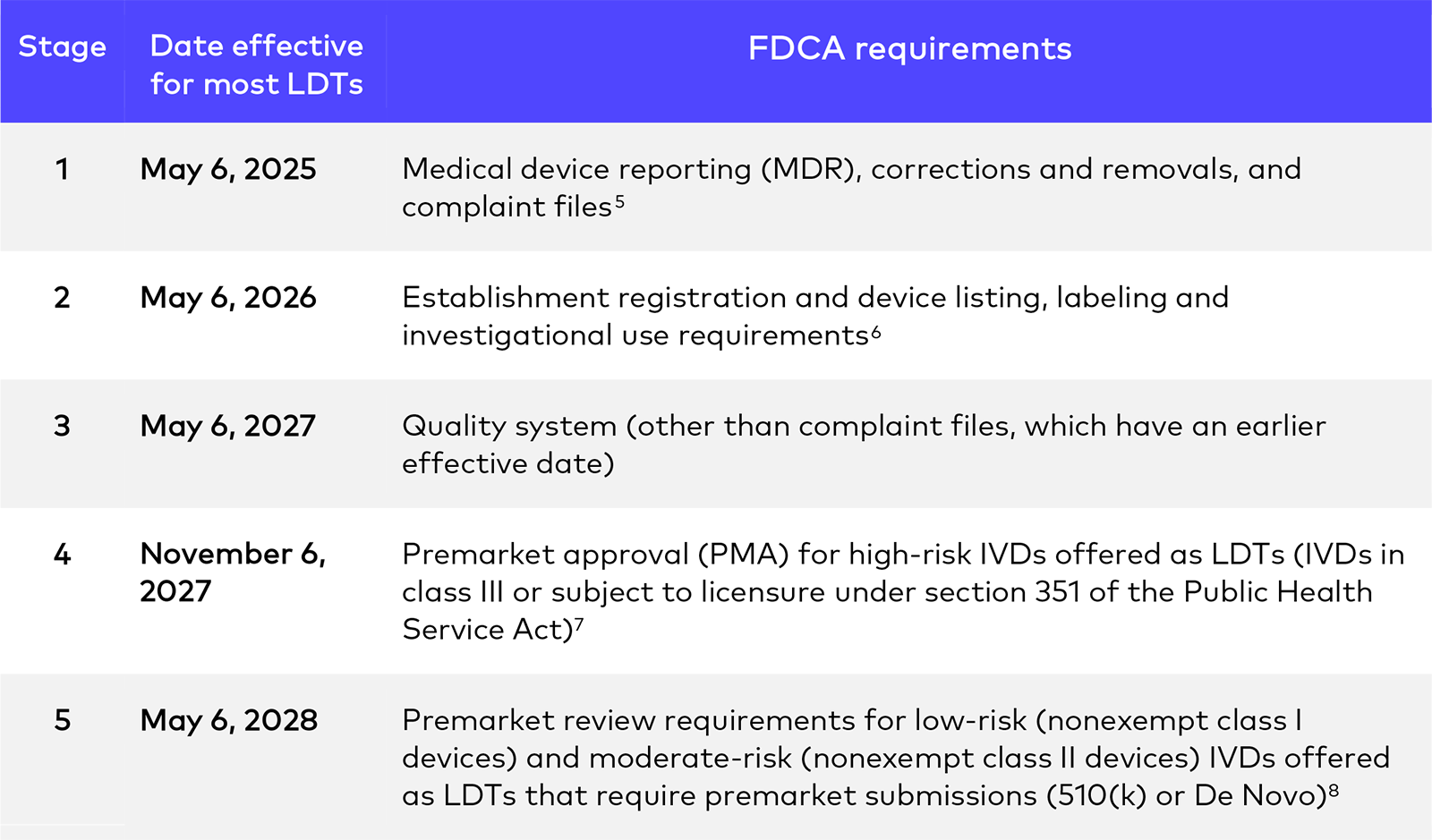
Thus, at the end of four years, FDA will generally expect FDCA compliance for the majority of LDTs, including companion diagnostics used in oncology.9 But the final rule also outlines certain categories of LDTs for which FDA will follow a different approach – requiring full and immediate FDCA compliance, continued exercise of full enforcement discretion, or a hybrid approach, namely, not requiring compliance with premarket review and quality system requirements except recordkeeping. This last category includes currently marketed LDTs that were first marketed before May 6, 2024. Such tests will be exempted from the new program, though they will have to comply with requirements other than premarket review and quality system requirements (except recordkeeping). 10
LDTs carved out from the four-year sunsetting

In addition, FDA does not intend to enforce premarket review requirements with respect to LDTs approved by the New York State Clinical Laboratory Evaluation Program, although these tests are still subject to requirements other than premarket review requirements.
Moreover, along with this final rule, FDA issued contemporaneously two new draft guidance documents to outline FDA’s policy on how it will approach tests responding to an “emergent situation,” such as the outbreak of an infectious disease or other public health emergencies.
A word about enforcement discretion
Enforcement discretion is not the same as a full carve out from FDA’s oversight, as we saw with certain categories of software in the 21st Century Cures Act. Thus, for all LDTs subject to enforcement discretion, it is important to remember that FDA retains jurisdiction over the products as medical devices, particularly in light of the final rule’s changes to the IVD definition. For a product subject to enforcement discretion, FDA may enforce the FDCA and pursue enforcement action for violations of the FDCA at any time it deems appropriate.
What comes next?
This final rule implements some of the policies the agency already had in place during the COVID-19 pandemic, such as requiring immediate compliance with the FDCA for tests intended for actual or potential emergencies or material threats declared under section 564 of the FDCA, and the continued exercise of enforcement discretion with respect to premarket review requirements for LDTs approved by the New York State Clinical Laboratory Evaluation Program. This enforcement discretion approach for the state of New York begs the question of whether other states were considered for this policy, or entities approved by other state clinical laboratory evaluation programs will challenge the rule.
As discussed in our November 2023 alert on the proposed rule, for this and other reasons, we anticipate litigation as FDA begins to enforce this new rule. Interestingly, the timing of this new rule coincides well with FDA’s rollout of the new Quality Management System Regulation (QMSR), which becomes effective on February 2, 2026. Therefore, LDT developers can focus on aligning their quality systems to the QMSR, without the uncertainty of whether it will be in effect when LDTs and LDT developers will be expected to comply with the quality system regulations.
Cooley senior regulatory analyst Kelly Marco also contributed to this alert.
Notes
- See Medical Devices; Laboratory Developed Tests, 88 FR at 68006 (October 3, 2023).
- 21 CFR § 809.3(a).
- Id.
- This policy applies to IVDs that are manufactured and offered as LDTs by laboratories that are certified under the Clinical Laboratory Improvement Amendments (CLIA), meet the regulatory requirements under CLIA to perform high-complexity testing, and used within such laboratories.
- See 21 CFR § 820.198.
- See 21 USC § 360 and 21 CFR Part 807 (establishment registration and device listing requirements); 21 USC § 352 and 21 CFR Parts 801 and 809 (labeling requirements); and 21 USC § 360j(g) and 21 CFR Part 812 (investigational use requirements).
- LDTs may continue to be offered after this date while a PMA is under review by FDA.
- LDTs may continue to be offered after this date while a 510(k) or De Novo is under review by FDA.
- See FDA’s June 2023 guidance, Oncology Drug Products Used with Certain In Vitro Diagnostics: Pilot Program.
- 21 CFR, Part 820, Subpart M.
- 21 CFR §§ 610.40, 1271.80(c).
- 21 CFR § 640.5.
- With the amendment, 21 CFR § 809.3(a) is amended as follows: “These products are devices as defined in section 201(h) of the [FDCA] and may also be biological products subject to section 351 of the Public Health Service Act, including when the manufacturer of these products is a laboratory.” (emphasis added)
Related Contacts


This content is provided for general informational purposes only, and your access or use of the content does not create an attorney-client relationship between you or your organization and Cooley LLP, Cooley (UK) LLP, or any other affiliated practice or entity (collectively referred to as "Cooley"). By accessing this content, you agree that the information provided does not constitute legal or other professional advice. This content is not a substitute for obtaining legal advice from a qualified attorney licensed in your jurisdiction, and you should not act or refrain from acting based on this content. This content may be changed without notice. It is not guaranteed to be complete, correct or up to date, and it may not reflect the most current legal developments. Prior results do not guarantee a similar outcome. Do not send any confidential information to Cooley, as we do not have any duty to keep any information you provide to us confidential. When advising companies, our attorney-client relationship is with the company, not with any individual. This content may have been generated with the assistance of artificial intelligence (Al) in accordance with our Al Principles, may be considered Attorney Advertising and is subject to our legal notices.